我室吕鑫教授在 ACS Catalysis 上发表论文:Hydrogenation of CO2 to Methane over a Ru/RuTiO2 Surface: A DFT Investigation into the Significant Role of the RuO2 Overlayer
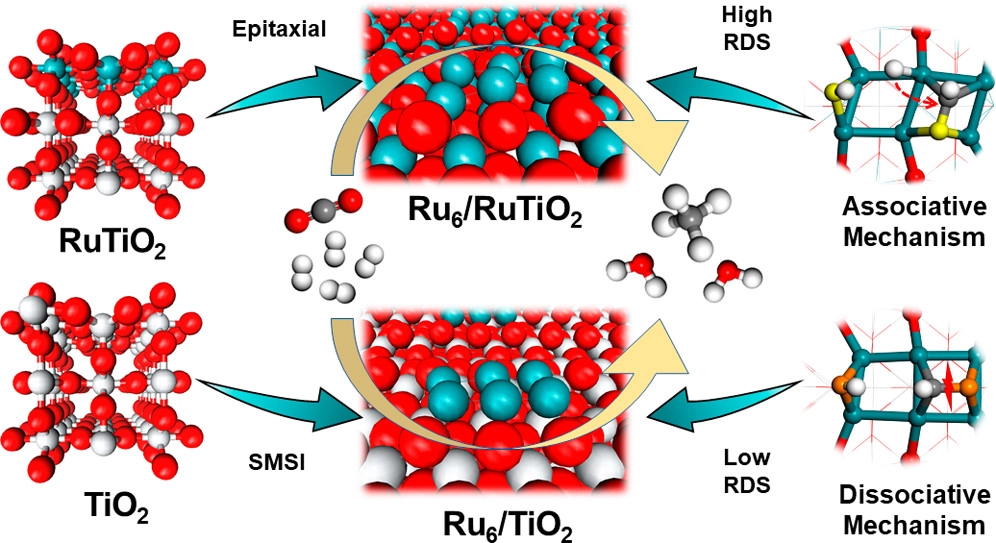
摘要:Understanding metal–support interactions is critical to the rational design of a catalyst for CO2 methanation. In this regard, the density functional theory is employed to shed light on the factors that determine the difference between RuTiO2- and TiO2-supported Ru nanoparticles. Structural observations and the calculated ratio of cohesive energy and adsorption energy (Ecoh/Eads) suggest that supported Ru6 could form as an epitaxial layer along with RuTiO2 as well as display strong metal–support interactions with TiO2 and thereby is used as a surface model to simulate the nanoparticles employed in the experiment. Furthermore, electronic analysis reveals that due to the existence of a RuO2 overlayer, more electrons are transferred from the support to the Ru cluster. Benefiting from this, CO2 is lower in adsorption energy since electrons from Ru6/RuTiO2 are less likely to fill in the antibonding orbital of Ru–O interaction. Analysis of the minimum-energy pathway indicates that the methanation of CO2 is led by C–O direct bond cleavage rather than the formate pathway in the first place for both surfaces, which is consistent with the FTIR spectroscopy results. Besides, we noticed that different reaction mechanisms control methane synthesis from the onset of CHO* formation. On the one hand, CHO* prefers an associative mechanism on Ru6/RuTiO2 due to the lower d-band center of the support and facile formation of CH2O* species. On the other hand, closer proximity of the d-band center to the Fermi level (EFermi) and preferable CH* formation promote CHO* on Ru6/TiO2 to undergo a dissociative pathway. Our comparative studies suggest that the RuO2 overlayer plays a key role in determining the reaction mechanism of CO2 methanation for Ru/r-TiO2.
文章链接:https://pubs.acs.org/doi/10.1021/acscatal.2c04539